La epidermólisis bullosa, más conocida como piel de mariposa, es una enfermedad incurable hoy en día, y cuya principal característica es la fragilidad de la piel y otros tejidos epiteliales del organismo.
En la actualidad afecta a aproximadamente 11 de cada millón de habitantes y 20 de cada millón de personas nacidas vivas, aunque estos datos varían en las distintas regiones geográficas del planeta.
Pero ¿cuál es la causa de esta enfermedad? ¿Qué posibilidades de tratamiento hay? Las últimas terapias génicas desarrolladas y ya aprobadas en Estados Unidos aportan una gran esperanza para el tratamiento de la enfermedad y constituyen un gran avance por su mecanismo para la creación de futuras terapias para este y otros trastornos.
No es la piel el único órgano afectado
La mayoría de los enfermos de piel de mariposa comparten signos y síntomas como la fragilidad e inflamación de la piel, la formación continua de ampollas o úlceras y dificultades en su cicatrización, lo que lleva en muchos casos a infecciones secundarias y complicaciones diversas.
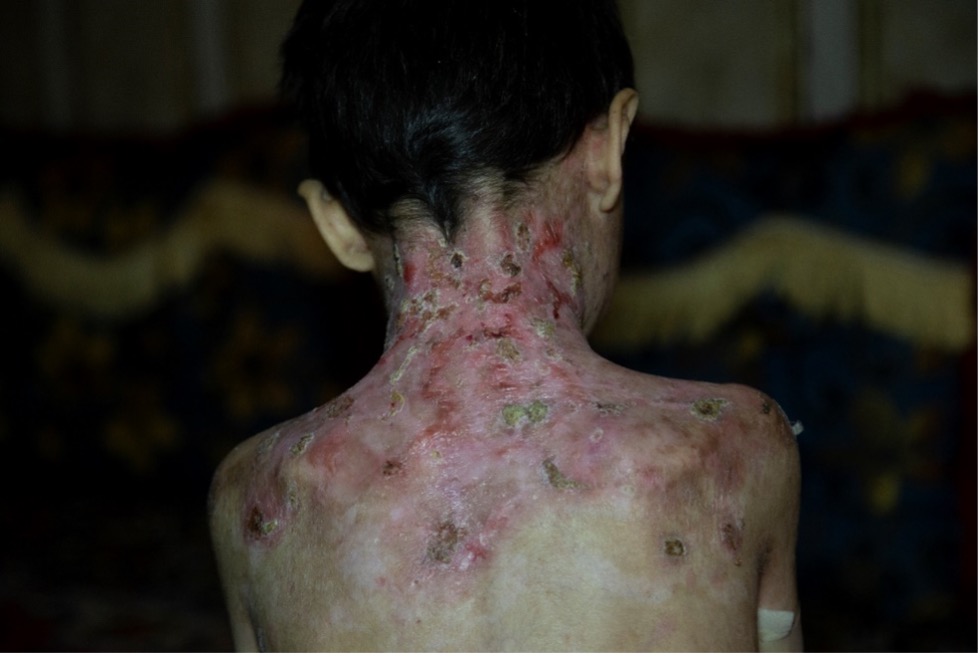
Efectos de la epidermólisis bullosa en la piel. Fuente: Wikipedia Commons
Dentro de estas complicaciones, la afectación del tejido epitelial de órganos y mucosas de todo el cuerpo puede acabar derivando en cardiopatías, fallos respiratorios, fallo renal, problemas de deglución por afectación del esófago y la faringe, problemas a lo largo del tracto gastrointestinal o problemas oculares, dentro de los que se ha reportado incluso ceguera. Sin olvidar la mayor complicación asociada a la piel de mariposa, el desarrollo de cáncer de células escamosas de la piel.
La inflamación o la fibrosis derivadas de la señalización molecular en una piel de mariposa crean un ambiente óptimo para el desarrollo de un tumor, provocando que la posibilidad de desarrollar un cáncer de células escamosas de la piel sea mucho mayor que en personas que no padecen la enfermedad.
Cuatro tipos de piel de mariposa
Los tratamientos actuales se centran en mejorar la vida de los pacientes, tratando de disminuir el picor, dolor o inflamación, factores que afectan gravemente al día a día de las personas que padecen la enfermedad. El objetivo utópico sería obtener una cura para esta enfermedad, pero en el camino se trata de que no tengan nada que envidiar en cuanto a su calidad de vida respecto a las personas que no padecen esta enfermedad.
Además de este tipo de tratamientos enfocados a calmar la sintomatología o las complicaciones, intentos de terapias génicas o terapias celulares, entre otras, se usan o están en camino de ello para tratar la piel de mariposa.
Pero es que el desarrollo de terapias se complica cuando se tiene en cuenta que dentro de lo que conocemos como piel de mariposa existen más de 30 trastornos, distintos por sus causas, características y posibles dianas terapéuticas. Hoy en día todos estos trastornos se clasifican en cuatro grandes grupos de piel de mariposa o epidermólisis bullosa: simple, juntural, distrófica o Kindler.
¿Y cuáles son las características que permiten distinguir estos cuatro tipos? Pues la clasificación se basa principalmente en los genes afectados. Es decir, cada uno de estos tipos de epidermólisis bullosa se produce por tener uno o varios genes no funcionales, los cuales permiten producir proteínas claves en la estructura del tejido epitelial.
El gen COL7A1 en la epidermólisis bullosa distrófica
Nos centraremos a partir de ahora en la epidermólisis bullosa distrófica, la más severa junto a la juntural. Su aparición se relaciona con la falta de una copia funcional del gen COL7A1, el cual permite producir colágeno de tipo VII. Esto es debido a mutaciones en el gen que hacen que no se pueda producir una proteína funcional.
Este colágeno de tipo VII forma las fibrillas de anclaje, que forman parte de la membrana basal, entre la epidermis y la dermis. Digamos que estas fibrillas de anclaje actúan como la unión entre la epidermis y la dermis, permitiendo que el tejido epitelial mantenga su estructura.
¿Qué ocurre entonces en la piel de las personas con epidermólisis bullosa? En el tejido epitelial de los enfermos las fibrillas de anclaje no pueden cumplir su función, lo que provoca una gran fragilidad a la estructura del tejido epitelial que forma nuestra piel, causando los síntomas propios de la enfermedad como ampollas, úlceras, dificultad para la cicatrización…
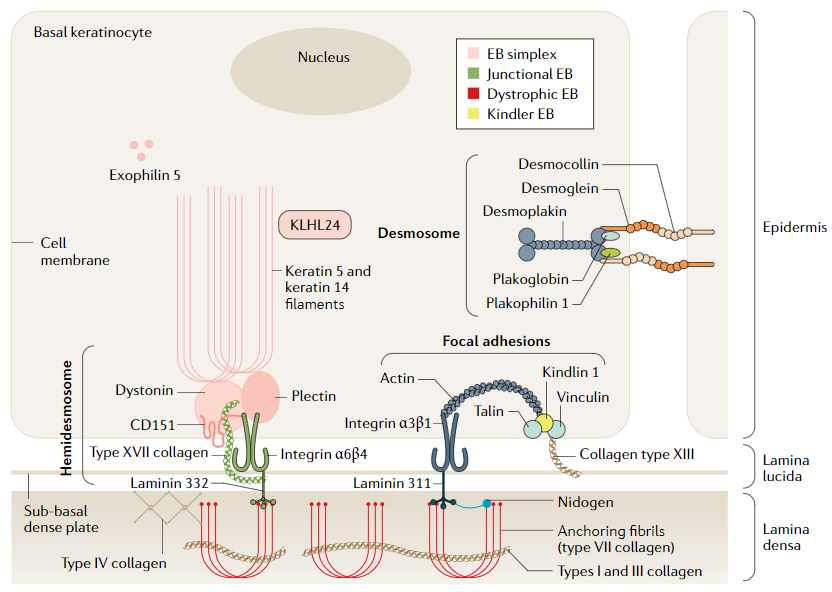
Esquema de la estructura del tejido epitelial y las proteínas afectadas la epidermólisis bullosa. Fuente: Bardhan, A. et al. (2020)
La solución “fácil” que se podría pensar sería proporcionar un gen COL7A1 funcional a las personas con piel de mariposa. Y eso es exactamente en lo que consiste la nueva terapia génica recientemente aprobada en Estados Unidos.
Una nueva terapia génica en forma de gel tópico
Básicamente el fundamento de la terapia, nombrada “beremagene gerperpavec” (B-VEC para hacerlo más sencillo) por los investigadores, es tratar de conseguir que los enfermos produzcan fibrillas de anclaje para que su tejido epitelial sea tan resistente como el de una persona no enferma. Y se está consiguiendo.
El método es medianamente sencillo de explicar. Dos copias funcionales del gen COL7A1 se introduce en un vector, en este caso en el virus del herpes simplex 1 (HSV-1), y se formula un gel tópico (que es en el caso de esta enfermedad lo más cómodo para los pacientes) que el paciente simplemente tiene que aplicarse en sus heridas, úlceras o ampollas. Se ha conseguido así la producción de colágeno tipo VII y de fibrillas de anclaje funcionales en enfermos de piel de mariposa, y se ha demostrado en ensayos clínicos una cicatrización del 67% de las heridas en 6 meses, aplicando el tratamiento cada 7 semanas aproximadamente.
Esquema de una partícula viral como con gen COL7A1. Fuente: Biorender.
En este caso se consiguió obtener un vector como el virus HSV-1 modificado para que no sea capaz de replicarse. Es decir, se modifica genéticamente el virus para que no sea capaz de reproducirse formando nuevos virus en nuestras células y por tanto infectarnos, pero que sí pueda hacerlo en unas condiciones determinadas en el laboratorio. De manera que en el laboratorio se replica la partícula viral con nuestro gen de interés en su interior, y una vez tenemos suficientes partículas, se fabrica el gel tópico que contiene las partículas. Una vez en la piel, el gen podrá ser adquirido y utilizado por nuestras células y el virus será incapaz de infectarnos porque no podrá tener descendencia.
Los grandes resultados de esta terapia génica han hecho que haya sido aceptada por la FDA (principal institución farmacéutica estadounidense) en mayo de este año, y que sea probablemente aceptada a lo largo del próximo año para su uso en Europa.
Conclusión
La creación de una terapia como esta es, además de una gran noticia para el tratamiento la epidermólisis bullosa distrófica, un paso más en el progreso de las terapias génicas y en su uso, cada vez más extendido, para tratar de forma específica distintas enfermedades con cierta base genética.
Bibliografía
- Bardhan, A., Bruckner-Tuderman, L., Chapple, I. L. C., Fine, J. D., Harper, N., Has, C., Magin, T. M., Marinkovich, M. P., Marshall, J. F., McGrath, J. A., Mellerio, J. E., Polson, R., & Heagerty, A. H. (2020). Epidermolysis bullosa. In Nature Reviews Disease Primers (Vol. 6, Issue 1). Nature Research. https://doi.org/10.1038/s41572-020-0210-0
- Hou, P. C., del Agua, N., Lwin, S. M., Hsu, C. K., & McGrath, J. A. (2023). Innovations in the Treatment of Dystrophic Epidermolysis Bullosa (DEB): Current Landscape and Prospects. In Therapeutics and Clinical Risk Management (Vol. 19, pp. 455–473). Dove Medical Press Ltd. https://doi.org/10.2147/TCRM.S386923
- Guide, S. v., Gonzalez, M. E., Bağcı, I. S., Agostini, B., Chen, H., Feeney, G., Steimer, M., Kapadia, B., Sridhar, K., Quesada Sanchez, L., Gonzalez, F., van Ligten, M., Parry, T. J., Chitra, S., Kammerman, L. A., Krishnan, S., & Marinkovich, M. P. (2022). Trial of Beremagene Geperpavec (B-VEC) for Dystrophic Epidermolysis Bullosa. New England Journal of Medicine, 387(24), 2211–2219. https://doi.org/10.1056/nejmoa2206663
- Gurevich, I., Agarwal, P., Zhang, P. P., Dolorito, J. A., Oliver, S., Liu, H., Reitze, N., Sarma, N., Bagci, I. S., Sridhar, K., Kakarla, V., Yenamandra, V. K., O’Malley, M., Prisco, M., Tufa, S. F., Keene, D. R., South, A. P., Krishnan, S. M., & Marinkovich, M. P. (2022). In vivo topical gene therapy for recessive dystrophic epidermolysis bullosa: a phase 1 and 2 trial. Nature Medicine, 28(4), 780–788. https://doi.org/10.1038/s41591-022-01737-y
- Lai-Cheong, J. E., McGrath, J. A., & Uitto, J. (2011). Revertant mosaicism in skin: Natural gene therapy. In Trends in Molecular Medicine (Vol. 17, Issue 3, pp. 140–148). https://doi.org/10.1016/j.molmed.2010.11.003
- Niti, A., Koliakos, G., & Michopoulou, A. (2023). Stem Cell Therapies for Epidermolysis Bullosa Treatment. In Bioengineering (Vol. 10, Issue 4). MDPI. https://doi.org/10.3390/bioengineering10040422
- FDA (19/05/2023). FDA Approves First Topical Gene Therapy for Treatment of Wounds in Patients with Dystrophic Epidermolysis Bullosa. https://www.fda.gov/news-events/press-announcements/fda-approves-first-topical-gene-therapy-treatment-wounds-patients-dystrophic-epidermolysis-bullosa

{kind=link}