

















La Química Computacional y la Bioinformática han adquirido un papel fundamental en el descubrimiento de fármacos y compuestos bioactivos. Estas disciplinas utilizan metodologías computacionales para buscar moléculas con propiedades específicas dentro de grandes librerías de compuestos. Sin embargo, estas técnicas son estáticas y no brindan información sobre la evolución temporal y los mecanismos moleculares involucrados. Para superar estas limitaciones, se recurre a la Dinámica Molecular (MD, por sus siglas en inglés), una técnica que permite simular y comprender el movimiento de las moléculas en detalle.
La Dinámica Molecular es ampliamente utilizada en el descubrimiento de fármacos y la Bioinformática Estructural. Esta técnica simula la dinámica de biomoléculas en disolución acuosa durante un periodo de tiempo determinado, explorando las diferentes conformaciones que pueden adoptar. Entre los diversos softwares disponibles para realizar este tipo de simulaciones, destaca GROMACS, un paquete de software libre y de código abierto muy popular.
El análisis posterior de las simulaciones de Dinámica Molecular es crucial para validar la simulación y obtener resultados relevantes. Permite identificar interacciones clave entre fármacos y proteínas diana, así como comprender la flexibilidad estructural de las proteínas al unirse a moléculas específicas. Sin embargo, el análisis de los archivos generados por GROMACS puede resultar complicado para usuarios sin experiencia en programación y línea de comandos. Por esta razón, hemos desarrollado una herramienta llamada ASGARD, que simplifica y automatiza el análisis de simulaciones de GROMACS.
ASGARD, una herramienta programada principalmente en Python y diseñada para sistemas Linux, permite a los usuarios realizar el análisis mediante una sola línea de comando, en lugar de tener que utilizar comandos individuales de GROMACS. Además, ASGARD incluye varios módulos que calculan aspectos esenciales de la simulación, como la estabilidad del sistema, la flexibilidad y las interacciones proteína-ligando. También genera automáticamente un informe que resume todos los análisis realizados, facilitando la comprensión de las interacciones y cambios estructurales en el sistema estudiado.
Para evaluar la utilidad de ASGARD, se realizaron pruebas utilizando los tutoriales proporcionados en el sitio web de GROMACS, los cuales describen los pasos para ejecutar una simulación de Dinámica Molecular en sistemas de lisozima en agua y el complejo lisozima-2-propilfenol.
Posteriormente, se simuló el sistema formado por la proteasa principal del SARS-CoV2 (MPro), involucrada en la replicación del virus, y el fármaco antiviral PaxlovidTM desarrollado por Pfizer. Después de esto, se analizaron las interacciones moleculares entre el fármaco y la proteína para comprobar la unión entre ambos. Estos casos de estudio demostraron que ASGARD es capaz de analizar automáticamente simulaciones de Dinámica Molecular en GROMACS, obteniendo resultados coherentes y generando un informe completo con los resultados obtenidos.
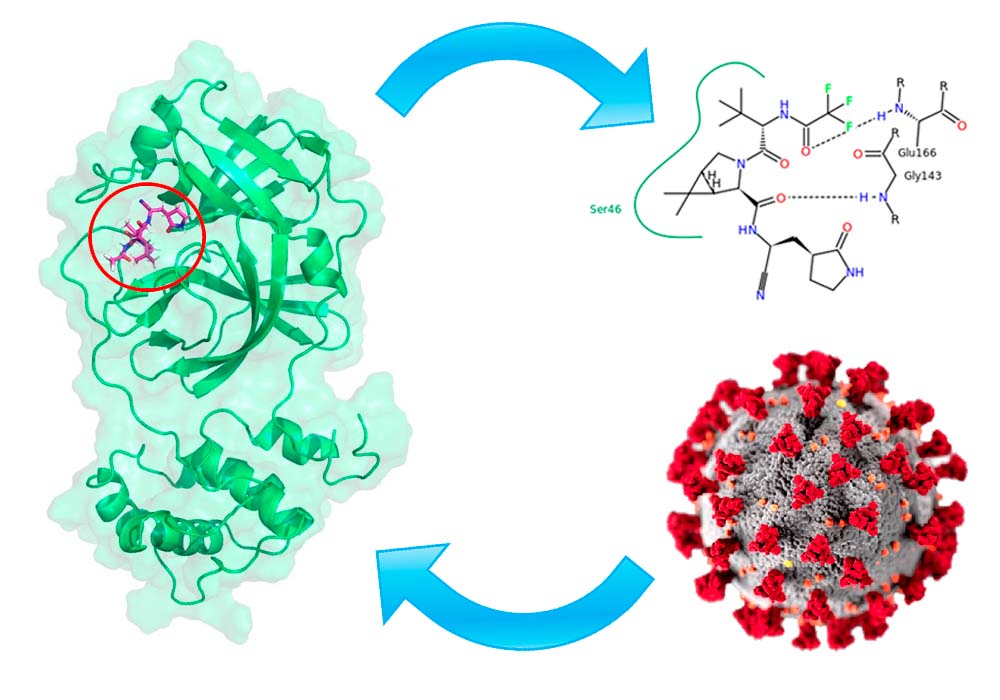
Al comparar los resultados obtenidos por ASGARD con datos experimentales en sistemas biológicos, se ha observado una notable correspondencia en términos de interacciones y estructura de proteínas. Los análisis de simulaciones de la lisozima en agua, el complejo lisozima-2-propilfenol y la proteasa principal del SARS-CoV2 con el fármaco antiviral PaxlovidTM han mostrado resultados consistentes con las observaciones experimentales previas. Estos hallazgos respaldan la fiabilidad y precisión de ASGARD como una herramienta para el análisis de simulaciones de Dinámica Molecular en GROMACS, brindando una comprensión concisa de las interacciones moleculares y los cambios estructurales en los sistemas estudiados.
En resumen, la combinación de la Química Computacional, la Bioinformática y la Dinámica Molecular ha revolucionado el campo del descubrimiento de fármacos. ASGARD, una herramienta desarrollada para simplificar el análisis de simulaciones de GROMACS, ha demostrado su eficacia al generar informes automáticos que permiten comprender las interacciones moleculares y los cambios estructurales en sistemas biológicos, como por ejemplo la unión de un fármaco con su diana o la evolución de un proceso molecular relacionado con una determinada enfermedad. De esta forma, esta herramienta promete facilitar la investigación en el descubrimiento de fármacos y la comprensión de procesos biológicos a nivel molecular.


